BuildingAndConstrainingMolecules
DASH solves structures by taking a model of the molecule (or molecules) in the asymmetric unit and moving it (them) around, subject to the constraints of space-group symmetry, until it finds a good match between calculated intensities and those derived from the Pawley fitting. If necessary, rotatable torsion angles in the molecule(s) are allowed to vary, either through a complete 360o range or through a smaller range defined by the user. If the asymmetric unit contains more than one molecule, they are moved independently of one another.
A requirement for solving structures, therefore, is to input appropriate 3D models of the molecule(s). Important things to consider are:
-
Building molecules in third-party programs (see Building Molecules in Third-Party Programs).
-
Converting molecules to Z-matrices (see Converting Molecules to Z-Matrix Format).
-
Reading molecules into DASH and defining the ranges through which bonds can be rotated (see Reading Molecules into DASH and Defining Rotatable Bonds).
-
Treatment of rings (see Treatment of Rings).
-
Treatment of stereoisomers (see Treatment of Stereoisomers).
-
Molecules on special positions (see Molecule Translation and Rotation; Special Positions).
-
Structures with more than one molecule in the asymmetric unit (see Structures with >1 Molecule per Asymmetric Unit).
There are several programs available for building 3D models of molecules, so DASH does not provide this capability (see Appendix C: Programs for Building 3D Molecules). Points to remember when building molecules are:
-
Ensure that the bond lengths, bond angles and ring conformations have reasonable values, since they will not normally be allowed to vary during simulated annealing. This is most easily achieved by using a fast force-field type minimisation in a modelling package.
-
Tables of standard bond lengths may be found in Volume C of International Tables for X-Ray Crystallography.
-
Torsion angles around single, acyclic bonds can usually be set to any value, since they will be varied during the simulated annealing process.
-
Stereochemistries will not be altered during simulated annealing, so if a molecule has more than one chiral centre, it is important that the relative stereochemistries are correct. (If you do not know what is correct, there may be no choice other than to try simulated annealing with each possibility in turn). Absolute stereochemistry does not matter.
-
The positions of hydrogen atoms will make little difference to fitting X-ray powder patterns, so protonation states and torsion angles involving H atoms are not critical. (In practice you may often omit H-atoms entirely.)
-
Write molecules out as mol or mol2 files. These can then be read into the DASH program for conversion into the Z-matrix format that DASH requires (see Using the Interface to Create Z-Matrix files).
-
Check the geometry of similar molecules in the CSD (see Using the Cambridge Structural Database (CSD) to Check Models).
-
Most molecular model building programs start from a user-created 2D diagram with bond types, from which to construct an approximate 3D model. This is then minimised from this starting point using various force-fields at whatever level of sophistication is available in the program.
-
For many molecules there will be no ambiguity as to the final 3D-model as regards the rigid portions, and the settings of any flexible torsion angles will not matter as DASH will recognise these and automatically set these as variable parameters in the structure solution search.
-
However, when ring systems are involved, or unusual combinations of elements in functional groups, the user is strongly advised to check for similar molecules in the Cambridge Structural Database (CSD), using the ConQuest search program. For example, this may reveal that a particular ring conformation is favoured in the experimental structures, and so one can adjust the 3D-molecular model accordingly.
-
It is worthwhile checking the bond lengths and bond angles for any unusual groups for significant deviation from the CSD average. It is probably wiser in such cases to construct the first trial model by taking accurate CSD values than to trust results from force-field energy minimisation. Indeed, for metal complexes the CSD examples are almost essential for good model building.
-
Torsion angle distributions may be easily obtained from the CSD using the ConQuest program, and searching on the appropriate fragments, or by using the direct link to Mogul, a molecular geometry database, from DASH (Mogul forms part of the CSD Portfolio which is available from the CCDC). The user may decide to reduce the flexibility of the model in DASH during the solution search by placing limits in the torsion angle ranges, or even fixing at certain values as in cases of intramolecular H-bonding.
-
In cases of ions such as chloride, there is much information in the CSD knowledge base, IsoStar, of intermolecular group…group interactions. This can be used in certain cases to predict the likely distance of an ion from a group in the main molecule, and can greatly improve the chances of solution.
-
H-bonding motifs may be important in certain structures with more than one molecule per asymmetric unit. It may be possible to find examples in CSD which would allow one with confidence to fix the relationship of the second molecule to the first by H-bonding, e.g. carboxylic acid centrosymmetric dimers, or chains with expected geometry.
In summary, use the CSD to check:
-
Bond lengths.
-
Bond angles.
-
Torsion angles ranges.
-
Ring conformations.
-
Small ions.
-
H-bond motifs (intra- and intermolecular).
-
Molecules built in third-party programs can be read by DASH and converted automatically into Z-matrix files (see Using the Interface to Create Z-Matrix files).
-
By default, the DASH will assign every single, acyclic bond as being rotatable (meaning that it will be varied during simulated annealing). This can be over-ridden, either by editing the Z-matrix file, or in DASH at the time of setting variable parameters for SA structure solution (see Checking and Setting Parameter Ranges).
-
Select Structure Solution either from the Mode menu, or by clicking the icon.
-
Select a .sdi file from the Molecular Z-Matrices window that appears by clicking on the Browse... button.
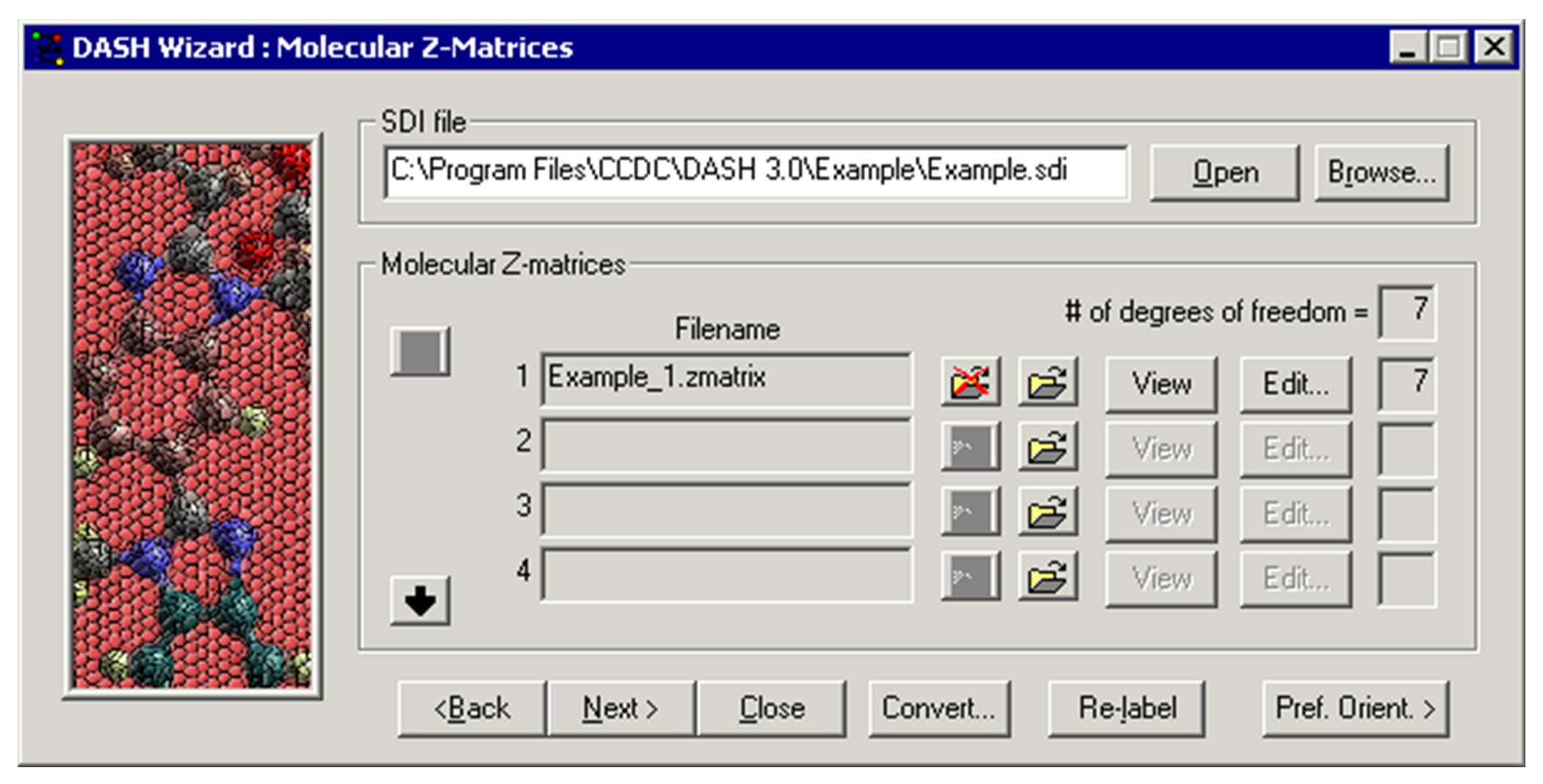
-
The allowed input formats for molecular model files are .res, .cssr, .pdb, .mol2, or .mol.
-
Click on the
 icon.
icon. -
Select from displayed files (in working directory).
-
Click Open.
-
This has created a file with extension .zmatrix which can then be used by DASH (see Reading Molecules into DASH and Defining Rotatable Bonds).
-
DASH reads molecules as Z-matrices. These can be created externally, or created internally by DASH when it reads a .mol or .mol2 file (see Converting Molecules to Z-Matrix Format).
-
The number of copies of a Z-matrix can be entered in the column labelled Number.
-
DASH automatically recognises all flexible torsion angles in the molecule for non-hydrogen atoms.
-
By default, the torsion angles around single, acyclic bonds will be varied through the full range of –180 to +180o during simulated annealing.
-
However, it is desirable to limit the number of variable parameters and their allowable ranges, since this will reduce the search space and increase the chances of structure solution. This is frequently possible with torsion angles. For example, a search of the Cambridge Structural Database shows that acyclic esters are invariably within 10o of the trans-conformation. Thus, the O=C-O-C torsion angle can be constrained to a range of, say, 10 to +10o, or even fixed at 0o.
-
Do not vary torsion angles that only affect the positions of H atoms, e.g. the torsion angles of OH, NH2 and CH3 groups. The data will not be sensitive to changes in these angles.
-
Many other torsion constraints can be inferred from the Cambridge Structural Database.
-
The best choice of constraints may depend on the quality of the data. For example, it is usually sensible to allow amides some flexibility by setting a range of –10 to +10o for the central torsion angle C-N-C=O. However, if the data is poor, it is probably better to fix the torsion at exactly zero.
-
It is sometimes useful to make repeated attempts at structure solution with torsion angles constrained to various likely ranges.
Rings can be handled in two ways in DASH:
-
You can input a likely ring conformation, obtained by looking at examples in the Cambridge Structural Database or by minimising in a modelling package, and keep the ring geometry fixed during simulated annealing. If the structure fails to solve, you can then try an alternative ring conformation.
-
You may have to postulate the positions of ring substituents too. For example, in the molecule below, 1,4-dichloro-1,4-dinitroso-cyclohexane, it is not only necessary to set the ring conformation (presumably chair) but also to decide whether the substituents are axial or equatorial:

- Alternatively, you can break one of the ring bonds and treat the resulting chain as a sequence of rotatable, acyclic torsion angles. This technique might be necessary if the ring is unusual and you have no idea about its probable conformation. However, it increases the number of variables significantly and also means that you are not taking advantage of the constraints imposed by ring closure. Thus, effectively, you are making the search space much larger.
- If a molecule has several possible stereoisomers, you may need to try simulated annealing with each in turn. e.g. cimetidine shown here, there are possible cis or trans positions of the CN group:
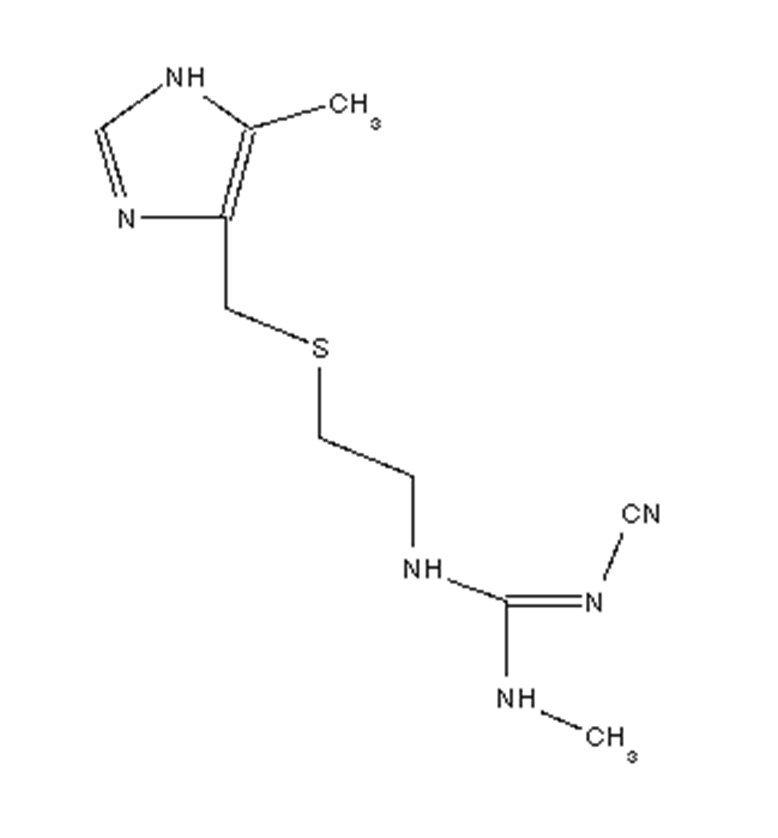
-
Sometimes, of course, you may be able to infer the probable stereochemistry from the chemical synthesis or from spectroscopic evidence.
-
You will not be able to determine absolute configuration from powder data.
-
Molecules will normally be allowed to translate and rotate freely in the unit cell, subject, of course, to the constraints of space group symmetry. This normally adds six degrees of freedom for each chemically discrete entity in the asymmetric unit, so solving structures with Z’>1 is much harder than solving structures with Z’=1.
-
Rotation is expressed as quaternion numbers Q0 – Q3. Rotations can be restricted to a single axis (see Editing Z-Matrix Rotations). There are four of these, but they are not mutually independent and actually contribute only three degrees of freedom to the problem (see Appendix I: References).
-
Fixed positions are sometimes required for molecules that occupy a special position in a space group. A common example is when a centrosymmetric molecule has its centre at the origin in a centrosymmetric spacegroup. This has to be handled by introducing a dummy atom into the Z-matrix. For example, a molecule can be constrained to sit on a centre of symmetry by including a dummy atom (of any element type but with a very low site occupation factor, e.g. 0.00001). This atom is positioned on the inversion centre (0.0, 0.0, 0.0) and anchored there by clicking the fixed box in the translation parameter list. A bond must be input from this dummy atom to any atom in the molecule, to allow the concept of the Z-matrix to be maintained. Rotations will still be allowed for this molecule, using this atom as the molecular origin reference point.
-
The easiest way to create the Z-matrix is to build a model with a model-building program, place a dummy atom at the centroid, and draw a bond to the nearest normal atom. The input this model file (mol2 or pdb format) in the normal way to the DASH Z-matrix conversion program. Then examine the Z-matrix file and edit the file to set this dummy atom to be the origin reference atom for the molecule (see Appendix H: Z-matrix format).
-
There are cases where one might want to specify a certain fixed distance to be maintained between a small molecule or ion; see the example in Structures with >1 Molecule per Asymmetric Unit.
If the structure contains more than one chemically-bonded unit (molecule or ion) in the asymmetric unit, each must be built separately and input to DASH. However, although you can vary the positions of more than one molecule or ion in the simulated annealing process, this has the disadvantage of significantly increasing the number of variables and so increases the complexity of the problem. Ways of avoiding this include:
-
Ignore one of the molecules: if you have two molecules, one of which is relatively small and so responsible for less than (say) 10% of the scattering, (i.e. only 10% or less of the sum of all the electrons in the asymmetric unit are in the smaller molecule), then DASH may be able to find a reasonable solution for just the larger residue. Typical examples would include leaving out water of crystallisation, or an ethanol molecule in presence of a large molecule like a steroid.
-
If this succeeds (i.e. produces a solution with a profile 2 slightly higher than that expected from a complete solution with all atoms present), the resulting model can be converted into a new Z-matrix. DASH can then be instructed to use the first atom in the molecule as an anchor point, all the torsion angles being constrained to the values found in the simulated annealing run. A second structure solution can then be attempted, optimising only the rotational orientation of the main molecule and both the position and the position and orientation of the small molecule. If certain H-bonds can be assumed to be present it may be possible in fact to tether a water molecule to be at a certain distance from a donor or acceptor atom on the main molecule.
-
Sometimes it is possible to guess the location of a small ion relative to a larger one. For example, in the following ion pair, it is highly likely that the chloride will be hydrogen bonded to the N-H group. Examination of the CSD database presented in IsoStar for an NH central group approached by a Cl- ion shows an average distance of 3.1 Å.
-
The method of constraining such an ion in the DASH SA procedure is best explained by this example. Using a model building program, construct a Cl atom at the required position relative to the N atom, draw a dummy-bond to the N atom, and output as a .mol2 or .pdb file. On reading into the DASH Z-matrix conversion program this produces a single Z-matrix file, where the Cl atom is now tethered to the N atom. The actual distance from N to Cl can of course be modified by directly editing the Z-matrix file, as can a dummy-bond angle. In this case the torsion angle involving Cl is not meaningful and can be set as fixed in the parameter list.