Tutorial05
Introduction
The object of this tutorial is to guide you through the structure solution of uridine. It assumes that you have completed Tutorial 1. In this tutorial, you will learn:
-
How to handle two molecules in the asymmetric unit
-
To appreciate the importance of a good molecular model.

Data
The data set Tutorial_5..xye is a synchrotron X-ray diffraction data set collected on BM16 at the ESRF, at 130K. The incident wavelength was 0.85075 Å.
Stage 1: Reading the Data
-
Open DASH and select the directory where the data resides.
-
Select View data / determine peak positions and click Next >.
-
Select the file Tutorial_5.xye using the Browse... button.
-
Click Next >.
-
Check that the wavelength and radiation source have been set correctly and click Next >.
Stage 2: Examining the Data
The data spans 4 to 30° 2θ. Truncate the data to 2.0 Å resolution. The background can be removed at this stage so proceed to do so. The default value for the window parameter of 100 is appropriate. When you are satisfied that the background fit (green line) is reasonable, click Apply and then Next >.
Stage 3. Fitting the Peaks to Determine the Exact Peak Positions
Select the first 23 peaks using the method described in Tutorial 1, Stage 3.
Here is a guide to the positions (° 2θ) of the first 23 peaks:
| 4.84 | 6.63 | 7.07 | 7.51 | 7.81 |
| 9.70 | 10.51 | 10.56 | 10.77 | 10.91 |
| 11.12 | 11.40 | 11.98 | 12.11 | 12.21 |
| 12.52 | 12.77 | 13.22 | 13.28 | 13.74 |
| 14.10 | 14.30 | 14.42 |
-
Click Next >.
-
Select Run> to run DICVOL or use another indexing program as described in Tutorial 1.
Stage 4. Indexing
If you have selected peaks which are very similar to those given in the previous stage the DICVOL program returns a monoclinic cell with a = 13.8703 Å, b = 14.7167 Å, c = 4.9207 Å, beta = 95.70° and cell volume = 999.47 Å3 with figures of merit M(23) = 74.8 and F(23) = 374.8. Other cells are suggested with beta greater than 96° but it is customary to choose the cell with the smallest angle. (This turns out to be in very good agreement with a single crystal structure reported in the Cambridge Structural Database [CSD] reference code BEURID10.)
Stage 5. Stop and Think
Does the cell make sense? In this case we estimate the molecular volume to be 17 x 15 Å3 (9C, 2N and 6O) + 12 x 5 Å3 (12 H) = 315 Å3. If there were 2 or 4 molecules in the unit cell we thus estimate volumes of 630 or 1260 Å3 respectively. The estimate for 4 molecules per cell is more likely, allowing for the fact that there is likely to be extensive H-bonding which will tend to make the cell smaller in volume.
Stage 6. Checking the Cell and Determining the Space Group
You should check through the space groups (scrolling through the choices with the arrow keys) until a good match between the tick marks and peak positions is obtained. A very good correspondence is achieved with space group P 1 21 1, number 4:b. Thus we need to attempt structure solution in P21 with 2 independent molecules in the asymmetric unit.
Stage 7. Extracting Intensities
Pick 8 peaks which are isolated using the method described in Tutorial 1, Stage 7. When 8 peaks have been chosen the Pawley Refinement Status window will pop up automatically. The initial 3 cycles of the least squares refinement only involves the terms corresponding to the background. This should give a Pawley χ2 of 3 or better, accept these three cycles. The next 5 cycles of the least squares refinement should bring the Pawley χ2 down to about 1.5. Accept your best Pawley fit, making a note of χ2 and save the file as Tutorial_5.sdi.
Stage 8. Molecule Construction
Construct a 3D molecular description of the molecule using your favourite modelling software and save it in pdb, mol or mol2 format. Care must be taken with the conformation of the ribose ring. The five-membered ribose ring is not planar - four atoms of the ring define a plane and the 5th atom will be found either above the plane (on the same side as the 6-membered heteronuclear ring) or below the plane. If we search the CSD for molecules very similar to uridine, we find that the torsion angle defined by O, C4’, C3’ and C2’ is either between 0 and +30° (C3’ lies below the plane) or C3’ lies above the plane (torsion angle between 0 and -40°. In uridine, C3’ lies above the plane and to start with, your molecular model should copy this. Later, you will run through the simulated annealing process with a molecular model of a different ring conformation. If you do not have access to a molecular modelling package a .mol2 file created by SPARTAN is included for the tutorial: Tutorial_5.mol2.
Stage 9. Setting up the Structure Solution Run
-
Continue on from the Pawley fitting stage by selecting Solve >.
-
Click on the
 icon and select Tutorial_5.mol2 (the file that you created in
Stage 8); a Z-matrix file called Tutorial__1.zmatrix will be
generated automatically.
icon and select Tutorial_5.mol2 (the file that you created in
Stage 8); a Z-matrix file called Tutorial__1.zmatrix will be
generated automatically. -
Read in the Tutorial_5_1.zmatrix file.
-
As there are two molecules in the asymmetric unit, read in the Tutorial_5.zmatrix file again.
At this point DASH will confirm that there are 16 independent parameters. These parameters are listed when you click on Next >. There are 3 parameters describing the positional co-ordinates, 4 (of which 3 independent) describing the molecular orientation within the unit cell and 2 variable torsion angles, for each molecule.
Click on Next >, leave the Simulated Annealing Protocol window with the default values, click Next > again and then click on Solve >. The structure should solve reasonably quickly.
Stage 10. Monitoring Structure Solution Progress
The progress of the structure solution can be followed by monitoring the profile χ2 and the difference plot. The molecule and crystal packing can be examined using the View button.
Stage 11. Examining the Output Structure
View the structure using the View button in the Results from Simulated Annealing window. All should look reasonable; there should be no abnormal close contacts between the atoms. In particular, check the formation of H-bonds. The picture below is taken from the Mercury visualiser for a SA good solution. In order to get a simple view of H-bonds we suggest clicking Show hydrogens off, and H-Bond on.

In the CSD it is found that in nearly every case H-bond donor atoms will be satisfied, so you should check all the OH groups, and the NH. Most of the O acceptor atoms will also take part in H-bonds, except for the ribose O which is often found not to accept. Note that in DASH the torsion angles involving H atoms are fixed by default at whatever value was input from the model - this means that the H-atoms do not necessarily point in the correct direction to form optimal H-bonds in these SA solutions.
For comparison an H-bond picture from Rpluto is given below for the single crystal structure BEURID10, which shows the same H-bond pattern. The directions of the axes may be inverted as absolute configuration cannot be determined from powder data. There is an ambiguity of 0.5 in the axial directions a and c, because of defined origin choice on a screw-axis, and the b-axis origin position is indeterminate.

Stage 12. Experiments with the Ring Conformation
When the molecular model was built, it was stressed that the ribose ring
is puckered, with the 3’ carbon out of the plane of the rest of the
ribose ring and on the same side as the 6-membered ring. Build a model
where the conformation of the ribose ring is different, for example
where the 3’ carbon of the ring points away from the 6 membered ring,
the other four atoms of the ring defining a plane. If you do not have
access to a model building package a file, Tutorial_5-2.mol2 is
included in the data files. Repeat the Simulated Annealing stage (you
can use DASH Wizard) but this time import Tutorial_5-2.mol2 and then
read in the newly created Tutorial_5-2_1.zmatrix file, as before.
Proceed with the solution stage. You will find that a good solution is
not found, and molecules may appear tangled, with close contacts. An
example result is shown below:
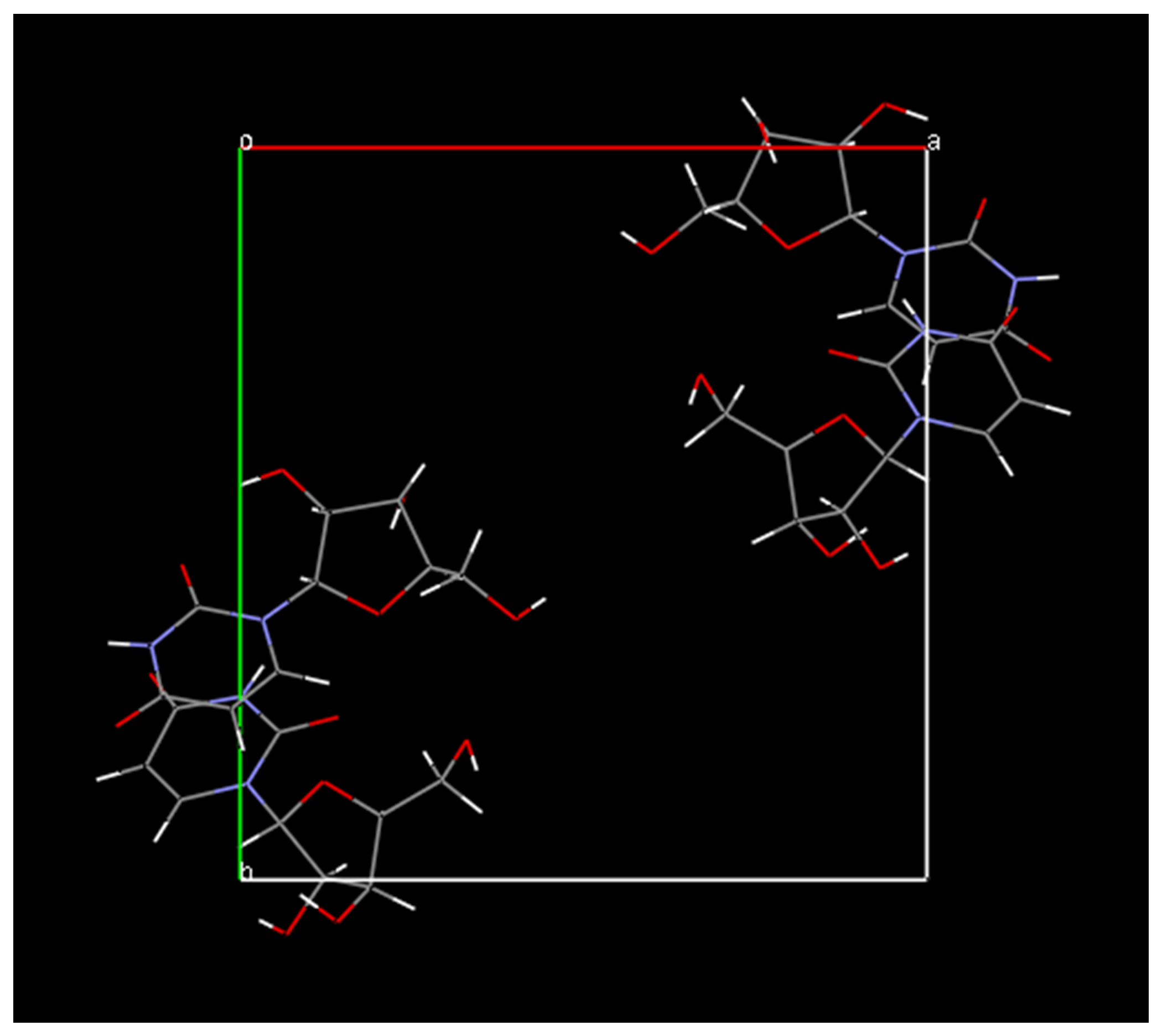
It should also be mentioned that you can experiment in DASH with flexible ring systems only by a rather crude method, whereby one takes the molecular model in the model building program and breaks a bond in the ribose ring - say C2’-C3’, and then export as a MOL2 file. Importing this into DASH then causes the rings to be treated as flexible chains, adding in fact 4 torsion angles per molecule; 8 torsion angles for the total SA search. This does not work in this case, as we have reached the present limit of the method with this data, but it is worth trying in less complex structures.
Stage 13. Conclusion
-
DASH can solve structures with two molecules per asymmetric unit.
-
The CSD should be consulted with regard to conformations of flexible rings.
-
Models with markedly wrong ring conformations will not give the correct solution.
References
*DICVOL Program:
*D. Louer & M. Louer (1972) J. Appl. Crystallogr. 5, 271-275.
A. Boultif & D. Louer (1991) J. Appl. Crystallogr. 24, 987-993.
*Model Builder:
*PC Spartan Pro Version 1.0.5 (16/8/2000) Copyright (1996-2000)
Wavefunction, Inc.
*Visualiser:
*Mercury (provided with DASH, has good H-bonding features)
*Single crystal structure (CSD reference code BEURID10):
*E.A. Green, R.D. Rosenstein, R. Shiono, D.J. Abraham, B.L. Trus, R.E.
Marsh (1975) Acta Crystallogr., B31, 102-107.